
Skip main navigation
×
Cookies Notification
We use cookies on this site to enhance your user experience. By continuing to browse the site, you consent to the use of our cookies.
Learn More

|
System Upgrade on Tue, May 28th, 2024 at 2am (EDT)
Existing users will be able to log into the site and access content. However, E-commerce and registration of new users may not be available for up to 12 hours.For online purchase, please visit us again. Contact us at customercare@wspc.com for any enquiries.

Series in Structural Biology: Volume 10
Single-Particle Cryo-Electron Microscopy
The Path Toward Atomic Resolution/ Selected Papers of Joachim Frank with Commentaries
Pages: 580- Edited by:
- Joachim Frank (Columbia University, USA)
Purchase
Save for later
Item saved, go to cart
ISBN: 978-981-323-485-7 (hardcover)
USD 209.00
Add to cart
ISBN: 978-981-323-487-1 (ebook)
USD 158.00
Add to cart
ISBN: 978-981-323-485-7 (hardcover)
Checkout
ISBN: 978-981-323-487-1 (ebook)
Checkout
The book reproduces 55 of more than 300 articles written by the author, representing milestones in methods development of single-particle cryo-EM as well as important results obtained by this technique in the study of biological macromolecules and their interactions. Importantly, neither symmetries nor ordered arrangements (as in two-dimensional crystals, helical assemblies, icosahedral viruses) are required. Although the biological applications are mainly in the area of ribosome structure and function, the elucidation of membrane channel structures and their activation and gating mechanisms are represented, as well. The book is introduced by a commentary that explains the original development of concepts, describes the contributions of the author's colleagues and students, and shows how challenges were overcome as the technique matured. Along the way, the ribosome served as an example for a macromolecule with intricate structure and conformational dynamics that pose challenges for three-dimensional visualization. Toward the end of the book — bringing us to the present time — molecular structures with near-atomic resolution are presented, and a novel type of computational analysis, manifold embedding, is introduced.
Single-particle cryo-EM is currently revolutionizing structural biology, presenting a powerful alternative to X-ray crystallography as a means to solve the structure of biological macromolecules. The book presents in one place a number of articles containing key advances in mathematical and computational methods leading up to the present time. Secondly, the development of the technique over the years is reflected by ever-expanding discoveries in the field of ribosome structure and function. Thirdly, as all histories of ideas, the history of concepts pertaining to this new method of visualization is fascinating all in itself.
Sample Chapter(s)
Chapter 1: Introduction and Commentaries
Contents:
- Image Formation, Optical Diffraction, and Digital Image Processing
- Development of Methods for 2D Averaging and 3D Reconstruction of Biological Macromolecules
- Focus Shifting from Methodology to Biology: the Ribosome and the Mechanism of Translation
- The "Resolution Revolution"
- Adding the Dimension of Time
- Outlook
- Acknowledgments
- Comprehensive List of Original Publications
Readership: Graduate students and researchers in biophysics, structural biology and protein synthesis.
Free Access
FRONT MATTER
- Pages:i–xii
https://doi.org/10.1142/9789813234864_fmatter
Preview Abstract
The following sections are included:
- Dedication
- Contents
Free Access
I. Introduction and Commentaries
- Pages:1–40
https://doi.org/10.1142/9789813234864_0001
Preview Abstract
As with many biologists trained as physicists, I entered biology through the “back door,” by initially focusing on developing or perfecting a tool, then demonstrating its utility in a particular area of biology, and finally allowing myself to be drawn into this area, after realizing that it was in fact possible to make useful contributions there. Reading Horace Freeland Judson’s book “The Eighth Day of Creation” (Judson, 1996) made me aware of the fact that I’m in good company, as Jim Watson and Francis Crick were complete outsiders when they started to approach one of the most vexing problems of biology…
II. Reprints of Selected Articles, in Chronological Order
No Access
Detection of Object Movement in the Optical Diffractograms of Electron Micrographs
- Pages:43–50
https://doi.org/10.1142/9789813234864_0002
Preview Abstract
Detection of object movement in the optical diffractograms of electron micrographs. Movements of the objects during an electron microscopic exposure cause characteristic modulations of the Fourier transform of the photographic density distribution, from which the nature of the movements can in turn be reconstructed. Modulations of this type were observed in optical diffractograms of a number of micrographs of amorphous carbon films and could be assigned to particular movements.
No Access
The Envelope of Electron Microscopic Transfer Functions for Partially Coherent Illumination
- Pages:51–68
https://doi.org/10.1142/9789813234864_0003
Preview Abstract
It is known that the linear transfer theory of optical image formation remains valid for partially coherent illumination, provided that the object contrast is small. Starting from the results of the theory of coherence, the properties of the amplitude and phase contrast transfer function are examined. For not too large source sizes, each transfer function can be represented as a product of the corresponding coherent transfer function and an envelope function. Such a product representation is convenient both for theoretical analysis and for the purpose of object restoration from images taken with partially coherent illumination. It is shown that the envelope function is closely related to the phase coherence function. The envelope functions are explicitly given for sources with disk-shaped and Gaussian intensity distributions. Electron microscopic image formation is then treated as a special case of the hitherto general analysis. The electron microscopic envelope functions are calculated for a Gaussian source, using generalized variables. In a second order approximation, the product representation can be retained, but it has to be modified to allow for a phase shift of the coherent transfer function. A comparison of exact and approximate computation of the partially coherent phase contrast transfer function shows that the formula derived in this paper works satisfactorily within the range of illumination apertures used in practice in high resolution experiments.
No Access
AVERAGING OF LOW EXPOSURE ELECTRON MICROGRAPHS OF NON-PERIODIC OBJECTS
- Pages:69–72
https://doi.org/10.1142/9789813234864_0004
Preview Abstract
The investigation concerns the possibility of extending to non-periodic objects the low exposure averaging techniques recently proposed for non-destructive electron microscopy of periodic biological objects. Two methods are discussed which are based on cross-correlation and are in principle suited for solving this problem.
No Access
Signal-to-noise ratio of electron micrographs obtained by cross correlation
- Pages:73–75
https://doi.org/10.1142/9789813234864_0005
Preview Abstract
The signal-to-noise ratio of electron micrographs can be determined by two-dimensional digital cross correlation even though neither signal nor noise can be analysed separately. Such measurements suggest how best to make use of the electron microscope.
No Access
MOTIF DETECTION IN QUANTUM NOISE-LIMITED ELECTRON MICROGRAPHS BY CROSS-CORRELATION
- W.O. SAXTON and
- J. FRANK
- Pages:76–84
https://doi.org/10.1142/9789813234864_0006
Preview Abstract
The use of cross-correlation to detect randomly positioned low-dose realizations of a motif in an image field is quantitatively evaluated. For both the bright- and dark-field cases, we derive theoretical expressions for the minimum dose allowing detection in terms of motif size, resolution, and contrast. Model computations on simulated low-dose images of a spherical virus particle give results that agree with our theoretical formulas and demonstrate the feasibility of this approach.
No Access
RECONSTRUCTION OF GLUTAMINE SYNTHETASE USING COMPUTER AVERAGING
- Pages:85–93
https://doi.org/10.1142/9789813234864_0007
Preview Abstract
The axial projection of the glutamine synthetase molecule has been reconstructed from electron micrographs of a stained preparation by using a new method of correlation search and averaging. The average over 50 individual molecules appears as a radial pattern with sixfold symmetry. The handedness evident in the average is attributed to nonuniformity of the negative stain.
No Access
SPIDER–A MODULAR SOFTWARE SYSTEM FOR ELECTRON IMAGE PROCESSING
- Pages:94–108
https://doi.org/10.1142/9789813234864_0008
Preview Abstract
The image-processing system SPIDER has been designed to operate on a minicomputer in a multiuser environment. SPIDER, which can be run either interactive or batch mode, makes a wide range of operations (including contrast enhancement, Fourier filtration, correlation averaging, and three-dimensional reconstruction) available for analysis of electron micrographs. The command language supports a hierarchical calling structure, branching commands, and DO-loops similar to those of FORTRAN.
No Access
Computer Averaging of Electron Micrographs of 40S Ribosomal Subunits
- Pages:109–111
https://doi.org/10.1142/9789813234864_0009
Preview Abstract
An enhanced lateral view of the 40S ribosomal subunit of HeLa cells has been obtained by computer averaging of single particles visualized in the electron microscope. Application of crystallographic criteria to independent averages shows that the reproducibility of the result is comparable to that obtained for thin, stained protein crystals by conventional Fourier filtration methods.
No Access
USE OF MULTIVARIATE STATISTICS IN ANALYSING THE IMAGES OF BIOLOGICAL MACROMOLECULES
- Pages:112–119
https://doi.org/10.1142/9789813234864_0010
Preview Abstract
We have developed a new technique of analysis that allows automatic classification of molecule images according to subtle differences. Computer alignment and multivariate statistical methods were used to analyze electron micrographic images of horseshoe crab hemocyanin half-molecules. The molecule projections fell into four distinct classes related to four different positions of the molecule on the grid. Averages obtained for each images subset are interpreted in terms of a three-dimensional model arrangement for the four subunits forming the half-molecule.
No Access
APPENDIX: Correspondence Analysis of Aligned Images of Biological Particles
- J. FRANK and
- M. VAN HEEL
- Pages:120–123
https://doi.org/10.1142/9789813234864_0011
Preview Abstract
The purpose of our analysis is to distinguish classes among the set of images, where each class is defined as a subset containing images more similar to each other than to images of any other subset. Differences in structure among the particles analyzed are reflected in differences in their projections. Conversely, if the angle of view is the same for all particles, then the existence of distinct classes in the particle images can be taken to indicate the presence of structural differences…
No Access
Reconstitution of molecule images analysed by correspondence analysis: a tool for structural interpretation
- Pages:124–137
https://doi.org/10.1142/9789813234864_0012
Preview Abstract
Correspondence analysis is gaining increasing importance in the analysis of electron micrographs of macromolecules. Partial or complete reconstitution of images from their factorial representations is introduced as a useful tool in interpreting variational patterns and tracing their physical origin. The value of image reconstitution is demonstrated with two examples, one using a set of model images and the other a set of images of a haemocyanin molecule assembly product.
No Access
Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli
- Pages:138–161
https://doi.org/10.1142/9789813234864_0013
Preview Abstract
We present a new reconstruction method that takes advantage of the fact that many biological macromolecular assemblies show a preferred orientation with respect to the plane of the specimen grid in the electron microscopic preparation. From one micrograph taken of such a specimen tilted by a large angle, a conical tilt series with random azimuthal angles can be extracted and used for a three-dimensional reconstruction. Our technique allows the determination of the molecular structure under low-dose conditions, which are not achievable with reconstruction methods that use conventional tilt series. The reconstruction method combines a number of existing image processing techniques with a newly developed weighted back-projection algorithm designed for three-dimensional reconstruction from projections taken with arbitrary projecting directions. The method is described as it was applied to the three-dimensional reconstruction of the structure of the 50S ribosomal subunit of Escherichia coli (E. coli).
No Access
Three-dimensional Structure of the Mammalian Cytoplasmic Ribosome
- Pages:162–174
https://doi.org/10.1142/9789813234864_0014
Preview Abstract
A three-dimensional reconstruction of the 80 S ribosome from rabbit reticulocytes has been calculated from low-dose electron micrographs of a negatively stained single-particle specimen. At 37 Å resolution, the precise orientations of the 40 S and 60 S subunits within the monosome can be discerned. The translational domain centered on the upper portion of the subunit/subunit interface is quite open, allowing considerable space between the subunits for interactions with the non-ribosomal macromolecules involved in protein synthesis. Further, the cytosolic side of the monosome is strikingly more open than the membrane-attachment side, suggesting a greater ease of communication with the cytoplasm, which would facilitate the inwards and outwards diffusion of a number of ligands. Although the 60 S subunit portion of the 80 S structure shows essentially all of the major morphological features identified for the eubacterial 50 S large subunit, it appears to possess a region of additional mass that evidently accounts for the more ellipsoidal form of the eukaryotic subunit.
No Access
Three-dimensional reconstruction of single particles embedded in ice
- Pages:175–195
https://doi.org/10.1142/9789813234864_0015
Preview Abstract
Single particles embedded in ice pose new challenges for image processing because of the intrinsically low signal-to-noise ratio of such particles in electron micrographs. We have developed new techniques that address some of these problems and have applied these techniques to electron micrographs of the Escherichia coli ribosome. Data collection and reconstruction follow the protocol of the random-conical technique of Radermacher et al. [J. Microscopy 146 (1987) 113]. A reference-free alignment algorithm has been developed to overcome the propensity of reference-based algorithms to reinforce the reference motif in very noisy situations. In addition, an iterative 3D reconstruction method based on a chi-square minimization constraint has been developed and tested. This algorithm tends to reduce the effects of the missing angular range on the reconstruction, thereby facilitating the merging of random-conical data sets obtained from differently oriented particles.
No Access
Eukaryotic Initiation Factor 3 Does Not Prevent Association Through Physical Blockage of the Ribosomal Subunit-Subunit Interface
- Pages:196–199
https://doi.org/10.1142/9789813234864_0016
Preview Abstract
The “native” 40 S ribosomal subunit, in which the protein eukaryotic initiation factor 3 is bound to the 40 S small ribosomal subunit, has been reconstructed to 48 Å resolution. Comparison with a previous three-dimensional reconstruction of the “derived” 40 S subunit lacking any non-ribosomal components reveals the attachment site and morphology of the factor. It is a large ( ∼ 165 to 170 Å long), bilobed, elongate structure, attached to the back lobes of the 40 S subunit by two strand-like features. Significantly, the factor is oriented away from the 60 S-subunit–40 S-subunit interface surface of the 40 S particle, suggesting that its anti-association activity is not accomplished via simple physical blockage of that surface.
No Access
The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles
- Pages:200–220
https://doi.org/10.1142/9789813234864_0017
Preview Abstract
Cryo-electron microscopy of single biological particles opens up new possibilities for structure analysis: the particle can be reconstructed in its native shape and internal features are preserved. To take advantage of these possibilities we have developed new methods of data collection and image processing and we have applied them to the 70S Escherichia coli ribosome. A method of orientation search is proposed, which makes it possible to relate random-conical data sets to one another even if they are collected from low-tilt micrographs. A technique for 3D alignment of projections is described and applied to the single-micrograph 3D reconstruction.
No Access
A model of protein synthesis based on cryo-electron microscopy of the E. coli ribosome
- Joachim Frank,
- Jun Zhu,
- Pawel Penczek,
- Yanhong Li,
- Suman Srivastava,
- Adriana Verschoor,
- Michael Radermacher,
- Robert Grassuccl,
- Ramani K. Lata, and
- Rajendra K. Agrawal
- Pages:221–224
https://doi.org/10.1142/9789813234864_0018
Preview Abstract
THE ribosome is formed by assembly of proteins and nucleic acids, and synthesizes proteins according to genetic instructions in all organisms. Many of the biochemical steps of this fundamental process are known, but a detailed understanding requires a well-defined structural model of the ribosome. Electron microscopy combined with image reconstruction of two-dimensional crystals or single ribosomes has been the most promising technique, but the resolution of the resulting models has been insufficient. Here we report a 25-Å reconstruction of the ribosome from Escherichia coli, obtained by combining 4,300 projections of ice-embedded single particles. Our new reconstruction reveals a channel in the small ribosomal subunit and a bifurcating tunnel in the large subunit which may constitute pathways for the incoming message and the nascent polypeptide chain, respectively. Based on these new findings, a three-dimensional model of the basic framework of protein synthesis is presented.
No Access
Direct Visualization of A-, P-, and E-Site Transfer RNAs in the Escherichia coli Ribosome
- Rajendra K. Agrawal,
- Pawel Penczek,
- Robert A. Grassucci,
- Yanhong Li,
- ArDean Leith,
- Knud H. Nierhaus, and
- Joachim Frank
- Pages:225–227
https://doi.org/10.1142/9789813234864_0019
Preview Abstract
Transfer RNA (tRNA) molecules play a crucial role in protein biosynthesis in all organisms. Their interactions with ribosomes mediate the translation of genetic messages into polypeptides. Three tRNAs bound to the Escherichia coli 70S ribosome were visualized directly with cryoelectron microscopy and three-dimensional reconstruction. The detailed arrangement of A- and P-site tRNAs inferred from this study allows localization of the sites for anticodon interaction and peptide bond formation on the ribosome.
No Access
Alignment of Conduits for the Nascent Polypeptide Chain in the Ribosome-Sec61 Complex
- Roland Beckmann,
- Doryen Bubeck,
- Robert Grassucci,
- Pawel Penczek,
- Adriana Verschoor,
- Günter Blobel, and
- Joachim Frank
- Pages:228–231
https://doi.org/10.1142/9789813234864_0020
Preview Abstract
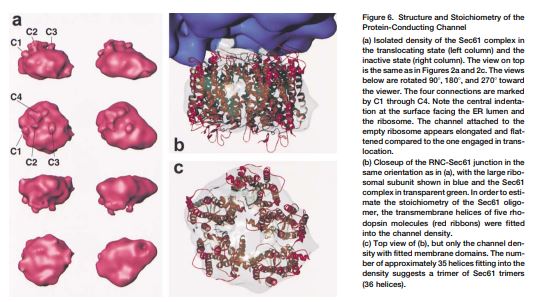
An oligomer of the Sec61 trimeric complex is thought to form the protein-conducting channel for protein transport across the endoplasmic reticulum. A purified yeast Sec61 complex bound to monomeric yeast ribosomes as an oligomer in a saturable fashion. Cryo–electron microscopy of the ribosome-Sec61 complex and a three-dimensional reconstruction showed that the Sec61 oligomer is attached to the large ribosomal subunit by a single connection. Moreover, a funnel-shaped pore in the Sec61 oligomer aligned with the exit of a tunnel traversing the large ribosomal subunit, strongly suggesting that both structures function together in the translocation of proteins across the endoplasmic reticulum membrane.
No Access
THREE DIMENSIONAL RECONSTRUCTION WITH CONTRAST TRANSFER COMPENSATION FROM DEFOCUS SERIES
- P.A. Penczek,
- J. Zhu,
- R. Schröder, and
- J. Frank
- Pages:232–239
https://doi.org/10.1142/9789813234864_0021
Preview Abstract
Cryo-electron microscopy provides the means to quantitatively study macromolecules in their native state. However, the original mass distribution of the macromolecule is distorted by the contrast transfer function (CTF) of the electron microscope. In addition, the zeros of the CTF put a practical limit on the resolution that can be achieved. Substantial improvement to the quality of the results can be accomplished by collecting the data using a series of defocus settings. Such data sets can be combined and the resolution can be extended beyond the first zero of the CTF. This procedure can be applied either at the stage of raw data, or more effectively at the stage of reconstructed volumes which have a high signal-to-noise ratio as a result of averaging over many projections. A method of three-dimensional (3D) reconstruction that combines an algebraic, iterative 3D reconstruction technique with CTF correction is proposed. The potential to incorporate a priori knowledge into the reconstruction process is discussed. This approach was used to obtain a 3D reconstruction of the E. coli 70S ribosome from energy-filtered cryo-images.
No Access
Visualization of elongation factor G on the Escherichia coli 70S ribosome: The mechanism of translocation
- Pages:240–244
https://doi.org/10.1142/9789813234864_0022
Preview Abstract
During protein synthesis, elongation factor G (EF-G) binds to the ribosome and promotes the step of translocation, a process in which tRNA moves from the A to the P site of the ribosome and the mRNA is advanced by one codon. By using three-dimensional cryo-electron microscopy, we have visualized EF-G in a ribosome-EF-G-GDP-fusidic acid complex. Fitting the crystal structure of EF-G-GDP into the cryo density map reveals a large conformational change mainly associated with domain IV, the domain that mimics the shape of the anticodon arm of the tRNA in the structurally homologous ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. The tip portion of this domain is found in a position that overlaps the anticodon arm of the A-site tRNA, whose position in the ribosome is known from a study of the pretranslocational complex, implying that EF-G displaces the A-site tRNA to the P site by physical interaction with the anticodon arm.
No Access
A 9 Å Resolution X-Ray Crystallographic Map of the Large Ribosomal Subunit
- Nenad Ban,
- Betty Freeborn,
- Poul Nissen,
- Pawel Penczek,
- Robert A. Grassucci,
- Robert Sweet,
- Joachim Frank,
- Peter B. Moore, and
- Thomas A. Steitz
- Pages:245–255
https://doi.org/10.1142/9789813234864_0023
Preview Abstract
The 50S subunit of the ribosome catalyzes the peptidyl-transferase reaction of protein synthesis. We have generated X-ray crystallographic electron density maps of the large ribosomal subunit from Haloarcula marismortui at various resolutions up to 9 Å using data from crystals that diffract to 3 Å. Positioning a 20 Å resolution EM image of these particles in the crystal lattice produced phases accurate enough to locate the bound heavy atoms in three derivatives using difference Fourier maps, thus demonstrating the correctness of the EM model and its placement in the unit cell. At 20 Å resolution, the X-ray map is similar to the EM map; however, at 9 Å it reveals long, continuous, but branched features whose shape, diameter, and right-handed twist are consistent with segments of double-helical RNA that crisscross the subunit.
No Access
Solution Structure of the E. coli 70S Ribosome at 11.5 Å Resolution
- Irene S. Gabashvili,
- Rajendra K. Agrawal,
- Christian M. T. Spahn,
- Robert A. Grassucci,
- Dmitri I. Svergun,
- Joachim Frank, and
- Pawel Penczek
- Pages:256–268
https://doi.org/10.1142/9789813234864_0024
Preview Abstract
Over 73,000 projections of the E. coli ribosome bound with formyl-methionyl initiator tRNA,Met were used to obtain an 11.5 Å cryo-electron microscopy map of the complex. This map allows identification of RNA helices, peripheral proteins, and intersubunit bridges. Comparison of double-stranded RNA regions and positions of proteins identified in both cryo-EM and X-ray maps indicates good overall agreement but points to rearrangements of ribosomal components required for the subunit association. Fitting of known components of the 50S stalk base region into the map defines the architecture of the GTPase-associated center and reveals a major change in the orientation of the α-sarcin-ricin loop. Analysis of the bridging connections between the subunits provides insight into the dynamic signaling mechanism between the ribosomal subunits.
No Access
A ratchet-like inter-subunit reorganization of the ribosome during translocation
- Pages:269–273
https://doi.org/10.1142/9789813234864_0025
Preview Abstract
The ribosome is a macromolecular assembly that is responsible for protein biosynthesis following genetic instructions in all organisms. It is composed of two unequal subunits: the smaller subunit binds messenger RNA and the anticodon end of transfer RNAs, and helps to decode the mRNA; and the larger subunit interacts with the amino-acid-carrying end of tRNAs and catalyses the formation of the peptide bonds. After peptide-bond formation, elongation factor G (EF-G) binds to the ribosome, triggering the translocation of peptidyl-tRNA from its aminoacyl site to the peptidyl site, and movement of mRNA by one codon. Here we analyse three-dimensional cryo-electron microscopy maps of the Escherichia coli 70S ribosome in various functional states, and show that both EF-G binding and subsequent GTP hydrolysis lead to ratchet-like rotations of the small 30S subunit relative to the large 50S subunit. Furthermore, our finding indicates a two-step mechanism of translocation: first, relative rotation of the subunits and opening of the mRNA channel following binding of GTP to EF-G; and second, advance of the mRNA(tRNA)2 complex in the direction of the rotation of the 30S subunit, following GTP hydrolysis.
No Access
Architecture of the Protein-Conducting Channel Associated with the Translating 80S Ribosome
- Roland Beckmann,
- Christian M.T. Spahn,
- Narayanan Eswar,
- Jürgen Helmers,
- Pawel A. Penczek,
- Andrej Sali,
- Joachim Frank, and
- Günter Blobel
- Pages:274–285
https://doi.org/10.1142/9789813234864_0026
Preview Abstract
In vitro assembled yeast ribosome-nascent chain complexes (RNCs) containing a signal sequence in the nascent chain were immunopurified and reconstituted with the purified protein-conducting channel (PCC) of yeast endoplasmic reticulum, the Sec61 complex. A cryo-EM reconstruction of the RNC-Sec61 complex at 15.4 Å resolution shows a tRNA in the P site. Distinct rRNA elements and proteins of the large ribosomal subunit form four connections with the PCC across a gap of about 10–20 Å. Binding of the PCC influences the position of the highly dynamic rRNA expansion segment 27. The RNC-bound Sec61 complex has a compact appearance and was estimated to be a trimer. We propose a binary model of cotranslational translocation entailing only two basic functional states of the translating ribosome-channel complex.
No Access
Structure of the 80S Ribosome from Saccharomyces cerevisiae–tRNA-Ribosome and Subunit-Subunit Interactions
- Christian M.T. Spahn,
- Roland Beckmann,
- Narayanan Eswar,
- Pawel A. Penczek,
- Andrej Sali,
- Günter Blobel, and
- Joachim Frank
- Pages:286–299
https://doi.org/10.1142/9789813234864_0027
Preview Abstract
A cryo-EM reconstruction of the translating yeast 80S ribosome was analyzed. Computationally separated rRNA and protein densities were used for docking of appropriately modified rRNA models and homology models of yeast ribosomal proteins. The core of the ribosome shows a remarkable degree of conservation. However, some significant differences in functionally important regions and dramatic changes in the periphery due to expansion segments and additional ribosomal proteins are evident. As in the bacterial ribosome, bridges between the subunits are mainly formed by RNA contacts. Four new bridges are present at the periphery. The position of the P site tRNA coincides precisely with its prokaryotic counterpart, with mainly rRNA contributing to its molecular environment. This analysis presents an exhaustive inventory of an eukaryotic ribosome at the molecular level.
No Access
Hepatitis C Virus IRES RNA–Induced Changes in the Conformation of the 40S Ribosomal Subunit
- Christian M. T. Spahn,
- Jeffrey S. Kieft,
- Robert A. Grassucci,
- Pawel A. Penczek,
- Kaihong Zhou,
- Jennifer A. Doudna, and
- Joachim Frank
- Pages:300–303
https://doi.org/10.1142/9789813234864_0028
Preview Abstract
Initiation of protein synthesis in eukaryotes requires recruitment of the 40S ribosomal subunit to the messenger RNA (mRNA). In most cases, this depends on recognition of a modified nucleotide cap on the 5’ end of the mRNA. However, an alternate pathway uses a structured RNA element in the 5’ untranslated region of the messenger or viral RNA called an internal ribosomal entry site (IRES). Here, we present a cryo-electron microscopy map of the hepatitis C virus (HCV) IRES bound to the 40S ribosomal subunit at about 20 Å resolution. IRES binding induces a pronounced conformational change in the 40S subunit and closes the mRNA binding cleft, suggesting a mechanism for IRES-mediated positioning of mRNA in the ribosomal decoding center.
No Access
Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process
- Mikel Valle,
- Jayati Sengupta,
- Neil K. Swami,
- Robert A. Grassucci,
- Nils Burkhardt,
- Knud H. Nierhaus,
- Rajendra K. Agrawal, and
- Joachim Frank
- Pages:304–317
https://doi.org/10.1142/9789813234864_0029
Preview Abstract
During the elongation cycle of protein biosynthesis, the specific amino acid coded for by the mRNA is delivered by a complex that is comprised of the cognate aminoacyl-tRNA, elongation factor Tu and GTP. As this ternary complex binds to the ribosome, the anticodon end of the tRNA reaches the decoding center in the 30S subunit. Here we present the cryo-electron microscopy (EM) study of an Escherichia coli 70S ribosome-bound ternary complex stalled with an antibiotic, kirromycin. In the cryo-EM map the anticodon arm of the tRNA presents a new conformation that appears to facilitate the initial codon–anticodon interaction. Furthermore, the elbow region of the tRNA is seen to contact the GTPase-associated center on the 50S subunit of the ribosome, suggesting an active role of the tRNA in the transmission of the signal prompting the GTP hydrolysis upon codon recognition.
No Access
Study of the Structural Dynamics of the E. coli 70S Ribosome Using Real-Space Refinement
- Haixiao Gao,
- Jayati Sengupta,
- Mikel Valle,
- Andrei Korostelev,
- Narayanan Eswar,
- Scott M. Stagg,
- Patrick Van Roey,
- Rajendra K. Agrawal,
- Stephen C. Harvey,
- Andrej Sali,
- Michael S. Chapman, and
- Joachim Frank
- Pages:318–330
https://doi.org/10.1142/9789813234864_0030
Preview Abstract
Cryo-EM density maps showing the 70S ribosome of E. coli in two different functional states related by a ratchet-like motion were analyzed using real-space refinement. Comparison of the two resulting atomic models shows that the ribosome changes from a compact structure to a looser one, coupled with the rearrangement of many of the proteins. Furthermore, in contrast to the unchanged inter-subunit bridges formed wholly by RNA, the bridges involving proteins undergo large conformational changes following the ratchet-like motion, suggesting an important role of ribosomal proteins in facilitating the dynamics of translation.
No Access
A cryo-electron microscopic study of ribosome-bound termination factor RF2
- Urmila B. S. Rawat,
- Andrey V. Zavialov,
- Jayati Sengupta,
- Mikel Valle,
- Robert A. Grassucci,
- Jamie Linde,
- Bente Vestergaard,
- Måns Ehrenberg, and
- Joachim Frank
- Pages:331–334
https://doi.org/10.1142/9789813234864_0031
Preview Abstract
Protein synthesis takes place on the ribosome, where genetic information carried by messenger RNA is translated into a sequence of amino acids. This process is terminated when a stop codon moves into the ribosomal decoding centre (DC) and is recognized by a class-1 release factor (RF). RFs have a conserved GGQ amino-acid motif, which is crucial for peptide release and is believed to interact directly with the peptidyl-transferase centre (PTC) of the 50S ribosomal subunit. Another conserved motif of RFs (SPF in RF2) has been proposed to interact directly with stop codons in the DC of the 30S subunit. The distance between the DC and PTC is ∼ 73 Å. However, in the X-ray structure ofRF2, SPF and GGQ are only 23 Å apart, indicating that they cannot be at DC and PTC simultaneously. Here we show that RF2 is in an open conformation when bound to the ribosome, allowing GGQ to reach the PTC while still allowing SPF– stop-codon interaction. The results indicate new interpretations of accuracy in termination, and have implications for how the presence of a stop codon in the DC is signalled to PTC.
No Access
Visualizing tmRNA Entry into a Stalled Ribosome
- Pages:335–338
https://doi.org/10.1142/9789813234864_0032
Preview Abstract
Bacterial ribosomes stalled on defective messenger RNAs (mRNAs) are rescued by tmRNA, an ∼300-nucleotide-long molecule that functions as both transfer RNA (tRNA) and mRNA. Translation then switches from the defective message to a short open reading frame on tmRNA that tags the defective nascent peptide chain for degradation. However, the mechanism by which tmRNA can enter and move through the ribosome is unknown. We present a cryo-electron microscopy study at ∼13 to 15 angstroms of the entry of tmRNA into the ribosome. The structure reveals how tmRNA could move through the ribosome despite its complicated topology and also suggests roles for proteins S1 and SmpB in the function of tmRNA.
No Access
Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy
- Mikel Valle,
- Andrey Zavialov,
- Wen Li,
- Scott M Stagg,
- Jayati Sengupta,
- Rikke C Nielsen,
- Poul Nissen,
- Stephen C Harvey,
- Måns Ehrenberg, and
- Joachim Frank
- Pages:339–346
https://doi.org/10.1142/9789813234864_0033
Preview Abstract
Aminoacyl-tRNAs (aa-tRNAs) are delivered to the ribosome as part of the ternary complex of aa-tRNA, elongation factor Tu (EF-Tu) and GTP. Here, we present a cryo-electron microscopy (cryo-EM) study, at a resolution of ∼9 Å, showing that during the incorporation of the aa-tRNA into the 70S ribosome of Escherichia coli, the flexibility of aa-tRNA allows the initial codon recognition and its accommodation into the ribosomal A site. In addition, a conformational change observed in the GTPase-associated center (GAC) of the ribosomal 50S subunit may provide the mechanism by which the ribosome promotes a relative movement of the aa-tRNA with respect to EF-Tu. This relative rearrangement seems to facilitate codon recognition by the incoming aa-tRNA, and to provide the codon-anticodon recognition-dependent signal for the GTPase activity of EF-Tu. From these new findings we propose a mechanism that can explain the sequence of events during the decoding of mRNA on the ribosome.
No Access
Locking and Unlocking of Ribosomal Motions
- Pages:347–358
https://doi.org/10.1142/9789813234864_0034
Preview Abstract
During the ribosomal translocation, the binding of elongation factor G (EF-G) to the pretranslocational ribosome leads to a ratchet-like rotation of the 30S subunit relative to the 50S subunit in the direction of the mRNA movement. By means of cryo-electron microscopy we observe that this rotation is accompanied by a 20 Å movement of the L 1 stalk of the 50S subunit, implying that this region is involved in the translocation of deacylated tRNAs from the P to the E site. These ribosomal motions can occur only when the P-site tRNA is deacylated. Prior to peptidyl-transfer to the A-site tRNA or peptide removal, the presence of the charged P-site tRNA locks the ribosome and prohibits both of these motions.
No Access
A twisted tRNA intermediate sets the threshold for decoding
- Pages:359–360
https://doi.org/10.1142/9789813234864_0035
Preview Abstract
Putting together consistent cryo-EM structure, transient kinetic and mutant tRNA suppressor data, it appears that a deformed or waggling aminoacyl-tRNA is the critical transitional structure examined by the ribosome during decoding at the A site. The unusual conformation may be required for effective proofreading of the codon–anticodon complex.
No Access
Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation
- Christian MT Spahn,
- Maria G Gomez-Lorenzo,
- Robert A Grassucci,
- Rene Jørgensen,
- Gregers R Andersen,
- Roland Beckmann,
- Pawel A Penczek,
- Juan PG Ballesta, and
- Joachim Frank
- Pages:361–372
https://doi.org/10.1142/9789813234864_0036
Preview Abstract
An 11.7-Å-resolution cryo-EM map of the yeast 80S · eEF2 complex in the presence of the antibiotic sordarin was interpreted in molecular terms, revealing large conformational changes within eEF2 and the 80S ribosome, including a rearrangement of the functionally important ribosomal intersubunit bridges. Sordarin positions domain III of eEF2 so that it can interact with the sarcinricin loop of 25S rRNA and protein rpS23 (Sl2p). This particular conformation explains the inhibitory action of sordarin and suggests that eEF2 is stalled on the 80S ribosome in a conformation that has similarities with the GTPase activation state. A ratchet-like subunit rearrangement (RSR) occurs in the 80S · eEF2 · sordarin complex that, in contrast to Escherichia coli 70S ribosomes, is also present in vacant 80S ribosomes. A model is suggested, according to which the RSR is part of a mechanism for moving the tRNAs during the translocation reaction.
No Access
The Cryo-EM Structure of a Translation Initiation Complex from Escherichia coli
- Pages:373–382
https://doi.org/10.1142/9789813234864_0037
Preview Abstract
The 70S ribosome and its complement of factors required for initiation of translation in E. coli were purified separately and reassembled in vitro with GDPNP, producing a stable initiation complex (IC) stalled after 70S assembly. We have obtained a cryo-EM reconstruction of the IC showing IF2•GDPNP at the intersubunit cleft of the 70S ribosome. IF2•GDPNP contacts the 30S and 50S subunits as well as fMet-tRNAfMet. IF2 here adopts a conformation radically different from that seen in the recent crystal structure of IF2. The C-terminal domain of IF2 binds to the singlestranded portion of fMet-tRNAfMet, thereby forcing the tRNA into a novel orientation at the P site. The GTP binding domain of IF2 binds to the GTPase-associated center of the 50S subunit in a manner similar to EF-G and EF-Tu. Additionally, we present evidence for the localization of IF1, IF3, one C-terminal domain of L7/L 12, and the N-terminal domain of IF2 in the initiation complex.
No Access
The structure of the 80S ribosome from Trypanosoma cruzi reveals unique rRNA components
- Pages:383–388
https://doi.org/10.1142/9789813234864_0038
Preview Abstract
We present analysis, by cryo-electron microscopy and single-particle reconstruction, of the structure of the 80S ribosome from Trypanosoma cruzi, the kinetoplastid protozoan pathogen that causes Chagas disease. The density map of the T. cruzi 805 ribosome shows the phylogenetically conserved eukaryotic rRNA core structure, together with distinctive structural features in both the small and large subunits. Remarkably, a previously undescribed helical structure appears in the small subunit in the vicinity of the mRNA exit channel. We propose that this rRNA structure likely participates in the recruitment of ribosome onto the 5′ end of mRNA, in facilitating and modulating the initiation of translation that is unique to the trypanosomes.
No Access
Estimation of variance in single-particle reconstruction using the bootstrap technique
- Pages:389–404
https://doi.org/10.1142/9789813234864_0039
Preview Abstract
Density maps of a molecule obtained by single-particle reconstruction from thousands of molecule projections exhibit strong changes in local definition and reproducibility, as a consequence of conformational variability of the molecule and non-stoichiometry of ligand binding. These changes complicate the interpretation of density maps in terms of molecular structure. A three-dimensional (3-D) variance map provides an effective tool to assess the structural definition in each volume element. In this work, the different contributions to the 3-D variance in a single-particle reconstruction are discussed, and an effective method for the estimation of the 3-D variance map is proposed, using a bootstrap technique of sampling. Computations with test data confirm the viability, computational efficiency, and accuracy of the method under conditions encountered in practical circumstances.
No Access
The process of mRNA–tRNA translocation
- Pages:405–412
https://doi.org/10.1142/9789813234864_0040
Preview Abstract
In the elongation cycle of translation, translocation is the process that advances the mRNA–tRNA moiety on the ribosome, to allow the next codon to move into the decoding center. New results obtained by cryoelectron microscopy, interpreted in the light of x-ray structures and kinetic data, allow us to develop a model of the molecular events during translocation.
No Access
Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization
- Sjors H W Scheres,
- Haixiao Gao,
- Mikel Valle,
- Gabor T Herman,
- Paul P B Eggermont,
- Joachim Frank, and
- Jose-Maria Carazo
- Pages:413–415
https://doi.org/10.1142/9789813234864_0041
Preview Abstract
Although three-dimensional electron microscopy (3D-EM) permits structural characterization of macromolecular assemblies in distinct functional states, the inability to classify projections from structurally heterogeneous samples has severely limited its application. We present a maximum likelihood—based classification method that does not depend on prior knowledge about the structural variability, and demonstrate its effectiveness for two macromolecular assemblies with different types of conformational variability: the Escherichia coli ribosome and Simian virus 40 (SV40) large T-antigen.
No Access
Visualization of the Hybrid State of tRNA Binding Promoted by Spontaneous Ratcheting of the Ribosome
- Xabier Agirrezabala,
- Jianlin Lei,
- Julie L. Brunelle,
- Rodrigo F. Ortiz-Meoz,
- Rachel Green, and
- Joachim Frank
- Pages:416–423
https://doi.org/10.1142/9789813234864_0042
Preview Abstract
A crucial step in translation is the translocation of tRNAs through the ribosome. In the transition from one canonical site to the other, the tRNAs acquire intermediate configurations, so-called hybrid states. At this stage, the small subunit is rotated with respect to the large subunit, and the anticodon stem loops reside in the A and P sites of the small subunit, while the acceptor ends interact with the P and E sites of the large subunit. In this work, by means of cryo-EM and particle classification procedures, we visualize the hybrid state of both A/P and P/E tRNAs in an authentic factor-free ribosome complex during translocation. In addition, we show how the repositioning of the tRNAs goes hand in hand with the change in the interplay between S13, L1 stalk, L5, H68, H69, and H38 that is caused by the ratcheting of the small subunit.
No Access
Exploration of parameters in cryo-EM leading to an improved density map of the E. coli ribosome
- Jamie LeBarron,
- Robert A. Grassucci,
- Tanvir R. Shaikh,
- William T. Baxter,
- Jayati Sengupta, and
- Joachim Frank
- Pages:424–432
https://doi.org/10.1142/9789813234864_0043
Preview Abstract
A number of image processing parameters in the 3D reconstruction of a ribosome complex from a cryo-EM data set were varied to test their effects on the final resolution. The parameters examined were pixel size, window size, and mode of Fourier amplitude enhancement at high spatial frequencies. In addition, the strategy of switching from large to small pixel size during angular refinement was explored. The relationship between resolution (in Fourier space) and the number of particles was observed to follow a lin-log dependence, a relationship that appears to hold for other data, as well. By optimizing the above parameters, and using a lin-log extrapolation to the full data set in the estimation of resolution from half-sets, we obtained a 3D map from 131,599 ribosome particles at 6.7 Å resolution (FSC = 0.5).
No Access
Flexible Fitting of Atomic Structures into Electron Microscopy Maps Using Molecular Dynamics
- Pages:433–443
https://doi.org/10.1142/9789813234864_0044
Preview Abstract
A novel method to flexibly fit atomic structures into electron microscopy (EM) maps using molecular dynamics simulations is presented. The simulations incorporate the EM data as an external potential added to the molecular dynamics force field, allowing all internal features present in the EM map to be used in the fitting process, while the model remains fully flexible and stereochemically correct. The molecular dynamics flexible fitting (MDFF) method is validated for available crystal structures of protein and RNA in different conformations; measures to assess and monitor the fitting process are introduced. The MDFF method is then used to obtain high-resolution structures of the E. coli ribosome in different functional states imaged by cryo-EM.
No Access
Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis
- Elizabeth Villa,
- Jayati Sengupta,
- Leonardo G. Trabuco,
- Jamie LeBarron,
- William T. Baxter,
- Tanvir R. Shaikh,
- Robert A. Grassucci,
- Poul Nissen,
- Måns Ehrenberg,
- Klaus Schulten, and
- Joachim Frank
- Pages:444–449
https://doi.org/10.1142/9789813234864_0045
Preview Abstract
In translation, elongation factor Tu (EF-Tu) molecules deliver aminoacyl-tRNAs to the mRNA-programmed ribosome. The GTPase activity of EF-Tu is triggered by ribosome-induced conformational changes of the factor that play a pivotal role in the selection of the cognate aminoacyl-tRNAs. We present a 6.7-Å cryo-electron microscopy map of the aminoacyl-tRNA·EF-Tu·GDP·kirromycin-bound Escherichia coli ribosome, together with an atomic model of the complex obtained through molecular dynamics flexible fitting. The model reveals the conformational changes in the conserved GTPase switch regions of EF-Tu that trigger hydrolysis of GTP, along with key interactions, including those between the sarcin-ricin loop and the P loop of EF-Tu, and between the effector loop of EF-Tu and a conserved region of the 16S rRNA. Our data suggest that GTP hydrolysis on EF-Tu is controlled through a hydrophobic gate mechanism.
No Access
Structural characterization of mRNA-tRNA translocation intermediates
- Xabier Agirrezabala,
- Hstau Y. Liao,
- Eduard Schreiner,
- Jie Fu,
- Rodrigo F. Ortiz-Meoz,
- Klaus Schulten,
- Rachel Green, and
- Joachim Frank
- Pages:450–455
https://doi.org/10.1142/9789813234864_0046
Preview Abstract
Cryo-EM analysis of a wild-type Escherichia coli pretranslocational sample has revealed the presence of previously unseen intermediate substates of the bacterial ribosome during the first phase of translocation, characterized by intermediate intersubunit rotations, L1 stalk positions, and tRNA configurations. Furthermore, we describe the domain rearrangements in quantitative terms, which has allowed us to characterize the processivity and coordination of the conformational reorganization of the ribosome, along with the associated changes in tRNA ribosome-binding configuration. The results are consistent with the view of the ribosome as a molecular machine employing Brownian motion to reach a functionally productive state via a series of substates with incremental changes in conformation.
No Access
High-resolution cryo-electron microscopy structure of the Trypanosoma brucei ribosome
- Yaser Hashem,
- Amedee des Georges,
- Jie Fu,
- Sarah N. Buss,
- Fabrice Jossinet,
- Amy Jobe,
- Qin Zhang,
- Hstau Y. Liao,
- Robert A. Grassucci,
- Chandrajit Bajaj,
- Eric Westhof,
- Susan Madison-Antenucci, and
- Joachim Frank
- Pages:456–462
https://doi.org/10.1142/9789813234864_0047
Preview Abstract
Ribosomes, the protein factories of living cells, translate genetic information carried by messenger RNAs into proteins, and are thus involved in virtually all aspects of cellular development and maintenance. The few available structures of the eukaryotic ribosome reveal that it is more complex than its prokaryotic counterpart, owing mainly to the presence of eukaryote-specific ribosomal proteins and additional ribosomal RNA insertions, called expansion segments. The structures also differ among species, partly in the size and arrangement of these expansion segments. Such differences are extreme in kinetoplastids, unicellular eukaryotic parasites often infectious to humans. Here we present a high-resolution cryo-electron microscopy structure of the ribosome of Trypanosoma brucei, the parasite that is transmitted by the tsetse fly and that causes African sleeping sickness. The atomic model reveals the unique features of this ribosome, characterized mainly by the presence of unusually large expansion segments and ribosomal-protein extensions leading to the formation of four additional inter-subunit bridges. We also find additional rRNA insertions, including one large rRNA domain that is not found in other eukaryotes. Furthermore, the structure reveals the five cleavage sites of the kinetoplastid large ribosomal subunit (LSU) rRNA chain, which is known to be cleaved uniquely into six pieces, and suggests that the cleavage is important for the maintenance of the T. brucei ribosome in the observed structure. We discuss several possible implications of the large rRNA expansion segments for the translation-regulation process. The structure could serve as a basis for future experiments aimed at understanding the functional importance of these kinetoplastid-specific ribosomal features in protein-translation regulation, an essential step towards finding effective and safe kinetoplastid-specific drugs.
No Access
Trajectories of the ribosome as a Brownian nanomachine
- Ali Dashti,
- Peter Schwander,
- Robert Langlois,
- Russell Fung,
- Wen Li,
- Ahmad Hosseinizadeh,
- Hstau Y. Liao,
- Jesper Pallesen,
- Gyanesh Sharma,
- Vera A. Stupina,
- Anne E. Simon,
- Jonathan D. Dinman,
- Joachim Frank, and
- Abbas Ourmazd
- Pages:463–475
https://doi.org/10.1142/9789813234864_0048
Preview Abstract
A Brownian machine, a tiny device buffeted by the random motions of molecules in the environment, is capable of exploiting these thermal motions for many of the conformational changes in its work cycle. Such machines are now thought to be ubiquitous, with the ribosome, a molecular machine responsible for protein synthesis, increasingly regarded as prototypical. Here we present a new analytical approach capable of determining the free-energy landscape and the continuous trajectories of molecular machines from a large number of snapshots obtained by cryogenic electron microscopy. We demonstrate this approach in the context of experimental cryogenic electron microscope images of a large ensemble of nontranslating ribosomes purified from yeast cells. The free-energy landscape is seen to contain a closed path of low energy, along which the ribosome exhibits conformational changes known to be associated with the elongation cycle. Our approach allows model-free quantitative analysis of the degrees of freedom and the energy landscape underlying continuous conformational changes in nanomachines, including those important for biological function.
No Access
Quantitative Connection between Ensemble Thermodynamics and Single-Molecule Kinetics: A Case Study Using Cryogenic Electron Microscopy and Single-Molecule Fluorescence Resonance Energy Transfer Investigations of the Ribosome
- Colin D. Kinz-Thompson,
- Ajeet K. Sharma,
- Joachim Frank,
- Ruben L. Gonzalez, Jr., and
- Debashish Chowdhury
- Pages:476–489
https://doi.org/10.1142/9789813234864_0049
Preview Abstract
At equilibrium, thermodynamic and kinetic information can be extracted from biomolecular energy landscapes by many techniques. However, while static, ensemble techniques yield thermodynamic data, often only dynamic, single-molecule techniques can yield the kinetic data that describe transition-state energy barriers. Here we present a generalized framework based upon dwell-time distributions that can be used to connect such static, ensemble techniques with dynamic, single-molecule techniques, and thus characterize energy landscapes to greater resolutions. We demonstrate the utility of this framework by applying it to cryogenic electron microscopy (cryo-EM) and single-molecule fluorescence resonance energy transfer (smFRET) studies of the bacterial ribosomal pre-translocation complex. Among other benefits, application of this framework to these data explains why two transient, intermediate conformations of the pre-translocation complex, which are observed in a cryo-EM study, may not be observed in several smFRET studies.
No Access
Activation of GTP hydrolysis in mRNA-tRNA translocation by elongation factor G
- Pages:490–496
https://doi.org/10.1142/9789813234864_0050
Preview Abstract
During protein synthesis, elongation of the polypeptide chain by each amino acid is followed by a translocation step in which mRNA and transfer RNA (tRNA) are advanced by one codon. This crucial step is catalyzed by elongation factor G (EF-G), a guanosine triphosphatase (GTPase), and accompanied by a rotation between the two ribosomal subunits. A mutant of EF-G, H91A, renders the factor impaired in guanosine triphosphate (GTP) hydrolysis and thereby stabilizes it on the ribosome. We use cryogenic electron microscopy (cryo-EM) at near-atomic resolution to investigate two complexes formed by EF-G H91A in its GTP state with the ribosome, distinguished by the presence or absence of the intersubunit rotation. Comparison of these two structures argues in favor of a direct role of the conserved histidine in the switch II loop of EF-G in GTPase activation, and explains why GTP hydrolysis cannot proceed with EF-G bound to the unrotated form of the ribosome.
No Access
Structural Basis for Gating and Activation of RyR1
- Amédée des Georges,
- Oliver B. Clarke,
- Ran Zalk,
- Qi Yuan,
- Kendall J. Condon,
- Robert A. Grassucci,
- Wayne A. Hendrickson,
- Andrew R. Marks, and
- Joachim Frank
- Pages:497–515
https://doi.org/10.1142/9789813234864_0051
Preview Abstract
The type-1 ryanodine receptor (RyR1) is an intracellular calcium(Ca2+) release channel required for skeletal muscle contraction. Here, we present cryo-EM reconstructions of RyR1 in multiple functional states revealing the structural basis of channel gating and ligand-dependent activation. Binding sites for the channel activators Ca2+, ATP, and caffeine were identified at interdomain interfaces of the C-terminal domain. Either ATP or Ca2+ alone induces conformational changes in the cytoplasmic assembly (“priming”), without pore dilation. In contrast, in the presence of all three activating ligands, high-resolution reconstructions of open and closed states of RyR1 were obtained from the same sample, enabling analyses of conformational changes associated with gating. Gating involves global conformational changes in the cytosolic assembly accompanied by local changes in the transmembrane domain, which include bending of the S6 transmembrane segment and consequent pore dilation, displacement, and deformation of the S4-S5 linker and conformational changes in the pseudo-voltage-sensor domain.
No Access
Key Intermediates in Ribosome Recycling Visualized by Time-Resolved Cryoelectron Microscopy
- Ziao Fu,
- Sandip Kaledhonkar,
- Anneli Borg,
- Ming Sun,
- Bo Chen,
- Robert A. Grassucci,
- Måns Ehrenberg, and
- Joachim Frank
- Pages:516–525
https://doi.org/10.1142/9789813234864_0052
Preview Abstract
Upon encountering a stop codon on mRNA, polypeptide synthesis on the ribosome is terminated by release factors, and the ribosome complex, still bound with mRNA and P-site-bound tRNA (post-termination complex, PostTC), is split into ribosomal subunits, ready for a new round of translational initiation. Separation of post-termination ribosomes into subunits, or “ribosome recycling,” is promoted by the joint action of ribosome-recycling factor (RRF) and elongation factor G (EF-G) in a guanosine triphosphate (GTP) hydrolysis-dependent manner. Here we used a mixing-spraying-based method of time-resolved cryo-electron microscopy (cryo-EM) to visualize the short-lived intermediates of the recycling process. The two complexes that contain (1) both RRF and EF-G bound to the PostTC or (2) deacylated tRNA bound to the 30S subunit are of particular interest. Our observations of the native form of these complexes demonstrate the strong potential of time-resolved cryo-EM for visualizing previously unobservable transient structures.
No Access
Structure and assembly model for the Trypanosoma cruzi 60S ribosomal subunit
- Zheng Liu,
- Cristina Gutierrez-Vargas,
- Jia Wei,
- Robert A. Grassucci,
- Madhumitha Ramesh,
- Noel Espina,
- Ming Sun,
- Beril Tutuncuoglu,
- Susan Madison-Antenucci,
- John L. Woolford, Jr.,
- Liang Tong, and
- Joachim Frank
- Pages:526–531
https://doi.org/10.1142/9789813234864_0053
Preview Abstract
Ribosomes of trypanosomatids, a family of protozoan parasites causing debilitating human diseases, possess multiply fragmented rRNAs that together are analogous to 28S rRNA, unusually large rRNA expansion segments, and r-protein variations compared with other eukaryotic ribosomes. To investigate the architecture of the trypanosomatid ribosomes, we determined the 2.5-Å structure of the Trypanosoma cruzi ribosome large subunit by single-particle cryo-EM. Examination of this structure and comparative analysis of the yeast ribosomal assembly pathway allowed us to develop a stepwise assembly model for the eight pieces of the large subunit rRNAs and a number of ancillary “glue” proteins. This model can be applied to the characterization of Trypanosoma brucei and Leishmania spp. ribosomes as well. Together with other details, our atomic-level structure may provide a foundation for structure-based design of antitrypanosome drugs.
No Access
Quantitative Characterization of Domain Motions in Molecular Machines
- Pages:532–541
https://doi.org/10.1142/9789813234864_0054
Preview Abstract
The work of molecular machines such as the ribosome is accompanied by conformational changes, often characterized by relative motions of their domains. The method we have developed seeks to quantify these motions in a general way, facilitating comparisons of results obtained by different researchers. Typically there are multiple snapshots of a structure in the form of pdb coordinates resulting from flexible fitting of low-resolution density maps, from X-ray crystallography, or from molecular dynamics simulation trajectories. Our objective is to characterize the motion of each domain as a coordinate transformation using moments of inertia tensor, a method we developed earlier. What has been missing until now are ancillary tools that make this task practical, general, and biologically informative. We have provided a comprehensive solution to this task with a set of tools implemented on the VMD platform. These tools address the need for reproducible segmentation of domains, and provide a generalized description of their motions using principal axes of inertia. Although this methodology has been specifically developed for studying ribosome motion, it is applicable to any molecular machine.
No Access
Channel opening and gating mechanism in AMPA-subtype glutamate receptors
- Edward C. Twomey,
- Maria V. Yelshanskaya,
- Robert A. Grassucci,
- Joachim Frank, and
- Alexander I. Sobolevsky
- Pages:542–558
https://doi.org/10.1142/9789813234864_0055
Preview Abstract
AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)-subtype ionotropic glutamate receptors mediate fast excitatory neurotransmission throughout the central nervous system. Gated by the neurotransmitter glutamate, AMPA receptors are critical for synaptic strength, and dysregulation of AMPA receptor-mediated signalling is linked to numerous neurological diseases. Here we use cryo-electron microscopy to solve the structures of AMPA receptor—auxiliary subunit complexes in the apo, antagonist- and agonist-bound states and determine the iris-like mechanism of ion channel opening. The ion channel selectivity filter is formed by the extended portions of the re-entrant M2 loops, while the helical portions of M2 contribute to extensive hydrophobic interfaces between AMPA receptor subunits in the ion channel. We show how the permeation pathway changes upon channel opening and identify conformational changes throughout the entire AMPA receptor that accompany activation and desensitization. Our findings provide a framework for understanding gating across the family of ionotropic glutamate receptors and the role of AMPA receptors in excitatory neurotransmission.
Free Access
BACK MATTER
- Pages:559–565
https://doi.org/10.1142/9789813234864_bmatter
Preview Abstract
The following section is included:
- Corpus Acknowledgments
Sample Chapter(s)
Chapter 1: Introduction and Commentaries






















